est2genome -space 2500000 -genome AC091491.fa -est NP105048.fa -align -outfile NP105048.AC091491.est2genome est2genome -space 2500000 -genome AC091491.fa -est THC1259113.fa -align -outfile THC1259113.AC091491.est2genome
spidey -i AC091491.fa -m NP105048.fa -p 0 > NP105048.AC091491.spidey spidey -i AC091491.fa -m THC1259113.fa -p 0 > THC1259113.AC091491.spidey
Have a careful look at these gene structures. Now, and in order to plot them, we transform both outputs to GFF format (General Feature Format. It's designed primarly for the exchange of gene prediction information). However, first we predict in these two cDNAs the coding region. In this way, we will be able to distinct coding exons from non-coding ones (bear in mind the structure of eukaryotic mRNAs). We will see now how to extract the Open Reading Frame (ORF) and the encoded protein from a cDNA.
Finding the encoded proteins: we run a gene prediction program to obtain the protein encoded in this cDNA. Connect to geneid, select geneid output and submit the sequence.
These are the predictions: NP105048.geneid and THC1259113.geneid
Are predictions what you expect? why don't we get a Single exon for both predictions? hint: think about signal prediction!
To solve this problem with the NP105048 cDNA, we decide to obtain all possible ORFs and keep the longest (usually the real one). Go to ORF Finder. Finding the ORF: Check the plot. to see in which strand and in which frame you get the longest ORF. Now translate the DNA sequence into protein:
FastaToTbl NP105048.fa | Translate 0 | TblToFasta > NP105048.prot.faTake a look at the program Translate, FastaToTbl and TblToFasta.
We could also have used this web server: Translate.
Finally, compare our predictions to the protein annotation for these cDNAs in the TIGR web site: NP105048 and THC1259113. Are they the same?
Apparently, coding regions span from nt:
- NP105048: 1 3828
- THC1259113: 82 3465
Now, we are ready to generate the gff files and plot them. For the est2genome output we add the UTR information.
- est2genome
est2genome2gff.pl -m 1 3828 NP105048.AC091491.est2genome | gawk '{$2="est2genome";$3="exon";$9="NP105048";print $0}' > NP105048.AC091491.est2genome.gff est2genome2gff.pl -m 82 3465 THC1259113.AC091491.est2genome | gawk '{$2="est2genome";$7="-";if ($3!="utr") $3="exon";$9="NP105048";print $0}' > THC1259113.AC091491.est2genome.gff
- spidey
gawk '$1=="Exon" && NF>6{gsub("[-%]"," "); print "NP105048","spidey","exon",$3,$4,$10,"+",".","NP105048"}' NP105048.AC091491.spidey > NP105048.AC091491.spidey.gff gawk '$1=="Exon" && NF>6{gsub("[-%]"," "); print "NP105048","spidey","exon",$4,$5,$11,"-",".","NP105048"}' THC1259113.AC091491.spidey | sort -k 4 -n > THC1259113.AC091491.spidey.gff
Compare gene structures, for both genes, output by these programs. Are they the same? Given that we know the THC1259113 gene is in reverse (check BLAST output) can you see any inconsistency in the raw est2genome output that leads to a wrong gff file? are coding and non-coding exons properly identified? hint: picture how corresponding positions should look like when aligning a genomic gene in reverse and a forward cDNA !
Let's fix this problem giving the CDS in relation to the reverse cDNA:
est2genome2gff.pl -m 693 4076 THC1259113.AC091491.est2genome | gawk '{$2="est2genome";$7="-";if ($3!="utr") $3="exon";$9="NP105048";print $0}' > THC1259113.AC091491.est2genome.rev.gff
We now plot the gene structures:
gff2ps -C gene.rc THC1259113.AC091491.est2genome.rev.gff THC1259113.AC091491.spidey.gff NP105048.AC091491.est2genome.gff NP105048.AC091491.spidey.gff > NP105048.THC1259113.AC091491.ps
We can convert the postscript output to gif (or jpeg) format, and include it within the html document.
convert -antialias -rotate 90 NP105048.THC1259113.AC091491.ps NP105048.THC1259113.AC091491.gif
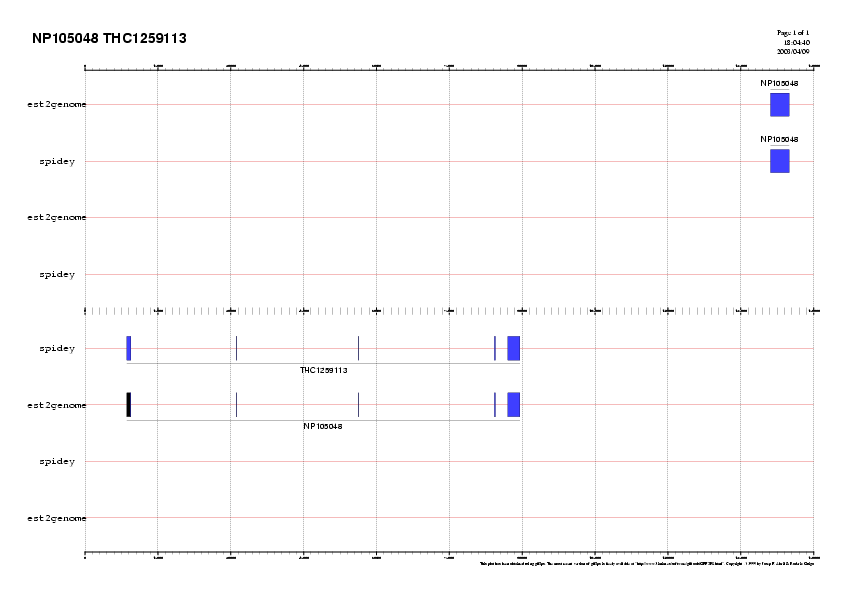
Note that UTRs appear in black.
Alignment of proteins
Now, we will define potential gene structures in our genomic region of interest, through the alignment of protein sequences. Previously, we have recovered the encoded proteins in two matching cDNAs to our genomic sequence and these are the amino acid sequences we will use. An alternative option would be blasting our genomic sequence against a set of known proteins, which is usually the case in the lab.We proceed now aligning proteins sequences against our genomic region of interest with two diferent protein-DNA alignment programs that take into account splice sites:
However, we will only use the genewise program:- genewise
genewise -genes -pretty -indel 1 NP105048.prot.fa AC091491.fa > NP105048.prot.AC091491.genewise genewise -genes -pretty -trev THC1259113.prot.fa AC091491.fa > THC1259113.prot.AC091491.genewise
How did we know that the THC1259113 protein is coded in reverse in the genomic AC091491 contig? hint: think of the BLAST output.
Be aware that, if needed, we could have used more distant protein homologs to build up the gene. However, we may have lost some accuracy.
We transform output for both genes to gff and plot them.
- genewise
gawk 'BEGIN{OFS="\t"}$1=="Exon" {print "AC091491","genewise",$1,start=($2<$3 ? $2 : $3), end=($3<$2 ? $2 : $2),".","+",".","NP105048"}' NP105048.prot.AC091491.genewise > NP105048.prot.AC091491.genewise.gff gawk 'BEGIN{OFS="\t"}$1=="Exon" {print "AC091491","genewise",$1,start=($2<$3 ? $2 : $3), end=($3<$2 ? $2 : $2),".","-",".","THC1259113"}' THC1259113.prot.AC091491.genewise > THC1259113.prot.AC091491.genewise.gff
gff2ps -C gene.rc THC1259113.prot.AC091491.genewise.gff NP105048.prot.AC091491.genewise.gff > THC1259113.NP105048.prot.AC091491.ps
We can convert the postscript output to gif (or jpeg) format, and include it within the html document.
convert -antialias -rotate 90 THC1259113.NP105048.prot.AC091491.ps THC1259113.NP105048.prot.AC091491.gif
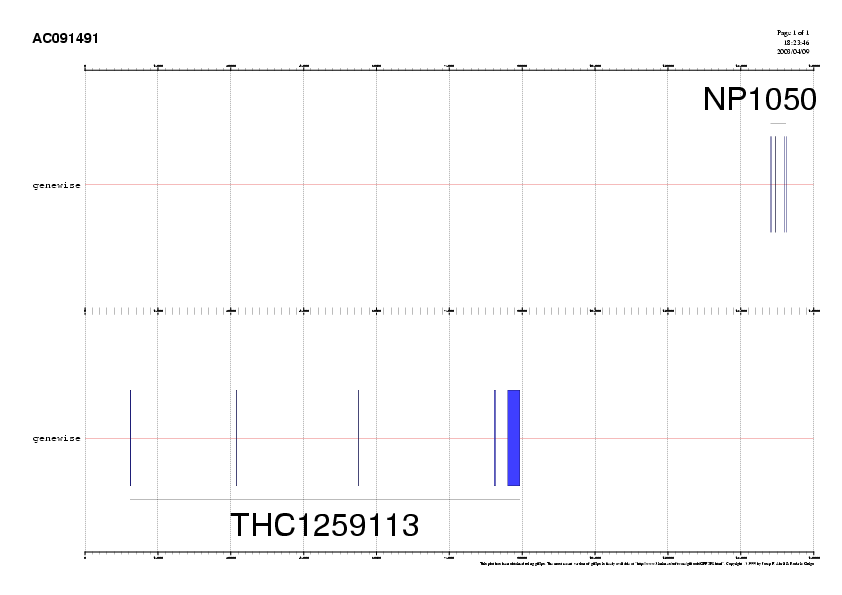
Compare these genes structures to the ones obtained previously by cDNA/DNA alignment. Are they the same? if not, why? (hint: try to plot them together) what is wrong with the NP105048 gene?
Gene prediction
We do now a gene prediction taking into account sequence similarity information. Connect to the a GenomeScan webserver. GenomeScan is a program for identifying the exon-intron structures of genes in genomic DNA sequences from a variety of organisms, with a focus on human and other vertebrates. The algorithm combines two principal sources of information:- Models of exon-intron and splice signal composition.
- Sequence similarity information such as BLASTX hits.
Check BLAST results: NP105048.prot.sw.homologs.blast and THC1259113.prot.sw.homologs.blast.
Save them in a fasta file: NP105048.prot.sw.homologs.fa and THC1259113.prot.sw.homologs.fa.
However, before running GenomeScan we will first mask the sequence in order to increase sensitivity (Sn) and specificity (Sp). A large fraction of genomic DNA belongs to relative small number of repeat families. A popular program to identify repeats in DNA sequences is RepeatMasker. There are a number of web servers which offer RepeatMasker analysis. We will use the EMBL RepeatMasker server.
RepeatMasker analysis produces a number of files. Among them:
By taking a look at this last file, we see that close to 40% of our sequence is made up of repetitive DNA.
It may be interesting to visualize the distribution of repeats along the sequence. There are a number of available tools to visualize genome annotations. We will use here gff2ps. There is a gff2ps web server at the pasteur institute, but the software can also be installed locally.
gff2ps requires the input file in gff format. The following awk script will do the conversion
grep AC091491 AC091491.seq.out | awk 'BEGIN{OFS="\t"}{print $5, $11, "repeat", $6,$7,".", ".", "."}' > AC091491.seq.out.gffNow we can run gff2ps:
gff2ps AC091491.seq.out.gff > AC091491.seq.out.pswhich we can visualize using ghostview:
gv AC091491.seq.out.psConcatenate protein similarity files:
cat NP105048.prot.sw.homologs.fa THC1259113.prot.sw.homologs.fa | sed '/^$/d' > NP105048.THC1259113.prot.sw.homologs.faNow we can run GenomeScan. Select the option: Predicted CDS and peptides.
Save results and check them: AC091491.masked.genomescan and AC091491.masked.genomescan.ps
We can convert the postscript output to gif (or jpeg) format, and include it within the html document.
convert -antialias -rotate 90 AC091491.masked.genomescan.ps AC091491.masked.genomescan.gif
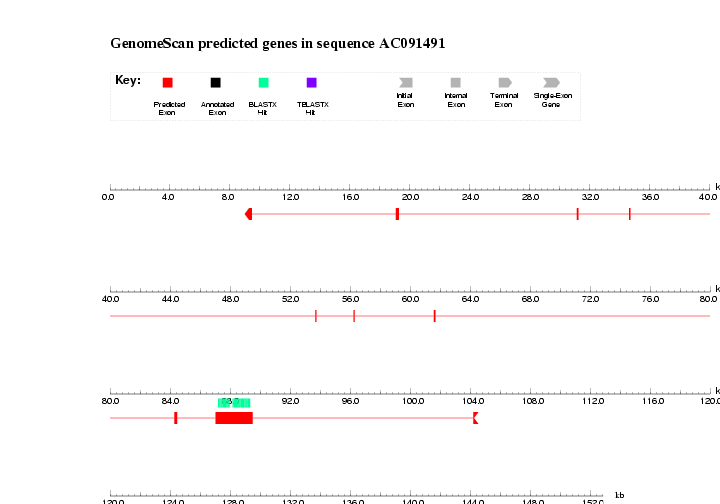
Do we get predictions for both genes? Is all similarity data used? If not, think why. Hint: check masked output from RepeatMasker.